相关文章
Win系统下安装
操作环境:
- python-2.7–window;
- mgltools_win32_1.5.6_Setup.exe;点击下载
- autodocksuite-4.2.6.i86Windows.exe;点击下载
- pymol-1.5.0.3.win32-py2.7(可视化);
- OpenBabel-3.0.0-x86.exe(pdbqt转pdb);
XiLock打包好的下载链接av65
安装步骤:
- 安装python
- 安装MGLTools1.5.6,路径无中文,完成后将其安装目录下的adt.bat创建快捷方式并置于工作目录,右键打开adt.bat的属性并将其“起始位置”更改为工作路径;
- 将autodocksuite-4.2.6.i86Windows.exe安装(该过程等于解压),将其中的autodock4.exe和autogrid4.exe复制到工作目录;
- pymol和OpenBabel正常安装即可。
Win系统下操作
准备工作
- 获取或建立receptor和ligand的pdb文件,去除水、不需要的杂原子;
- 双击adt.bat启动AutoDockTools;
Ligand
- Ligand>Input>Open打开ligand的pdb,软件会提示结构中的非极性氢原子、芳香碳原子个数、可旋转建等信息,点击确定即可;
- Ligand>Input>Choose,选择相应的ligand分子;
- Ligand>Output>Save as PDBQT,保存成为pdbqt文件,该文件中包含了配体结构中的原子信息和可旋转建的信息等;
注意:
DNA和RNA做ligand时要重新处理原子名称,在载入前将原子名中的单引号去掉,等对接完导出后再加上,否则可能识别不出来。
Grid
- Grid>Macromolecule>Open打开receptor的pdb文件,在左侧Dashboard窗口的选择方框中把受体蛋白勾选上,此时蛋白会变黄,即选中状态;点击Edit>Hydrogens>Add,ADT会为蛋白质加氢(由于解析技术的原因,氨基酸的氢原子在晶体结构中是不存在的,因此需要手动加氢原子)。如果第一步预处理的时候已经用其他软件加过H了,这里就不需要了。
- Grid>Macromolecule>Choose选择receptor分子,件提示结构中包含的非键原子、电荷等信息,点击确定,软件会自动弹出保存对话框,将受体保存成pdbqt文件。;
- Grid>Set Map Types>Choose Ligand,选择相应ligand分子;
- Grid>Grid Box,设置对接的盒子大小、坐标、格点数、格点距离,这一步需要自己根据不同的结构来进行具体确认。
- File>Close saving current保存盒子信息,选择Grid>Output>Save GPF,保存为protein_ligand.gpf文件;
Docking参数设置
- Docking>Macromolecule>Set Rigid Filename,选择receptor的pdbqt文件,将受体蛋白质设置为刚性;
- Docking>Ligand>Choose,选择配体,设置初始位置等信息,点击Accept。
- Docking>Output>Lamarckian GA(4.2),选择拉马克遗传算法作为对接算法,保存成为protein_ligand.dpf文件。dpf文件中包含了分子对接的参数,默认对接的构象数为10个,可以手动修改对接的构象数目(倒数第二行的ga_run 10修改为自己想要的数值);
Run
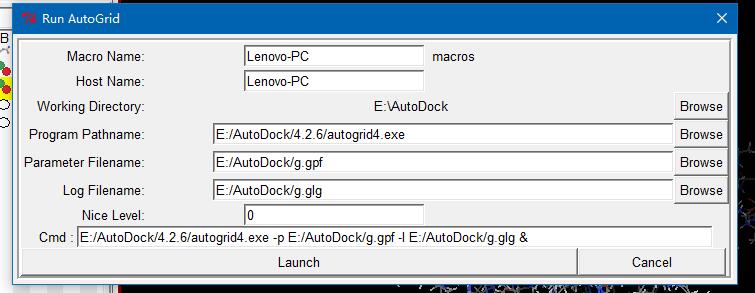
- Run->Run Autogrid,在Log Filename和Cmd的“-l”后输入以.glg为后缀的文件名,Launch后有个小弹窗出来,等待它消失就好了,此时工作目录会多出一堆文件;
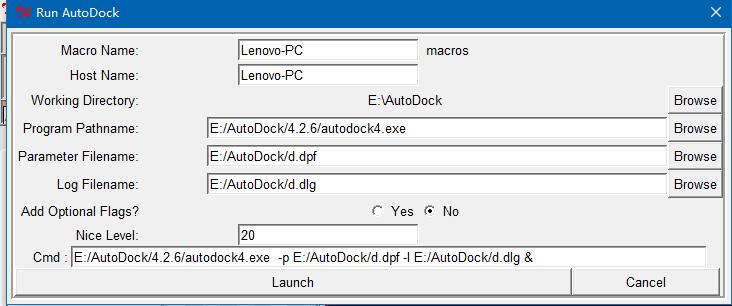
- Run->Run AutoDock,在Log Filename和Cmd的“-l”后输入以.dlg为后缀的文件名,Launch后有个小弹窗出来,等待它消失,发现多了一个dlg文件,这就是对接的最终结果,一般有10种对接方式。;
结果分析
- Analyze->Open,打开刚才生成的.dlg文件;
- Analyze->Macromolecule->Open,加上受体大分子;
- Analyze->Conformations->Play,一个个的查看刚才对接的结果;
- 导出对接结果并转换成PDB格式的数据时,点击像“&”符号的按钮,然后再点击“Write Complex”按钮生成pdbqt文件,(命名时注意不要忘了后缀名)。打开Open Babel软件,找到刚才生成的pdbqt文件,以及写好输出的pdb文件名,然后点击CONVERT按钮。
参考资料:
Q&A
-
[分子对接教程 (7) AutoDock对接中易错问题](https://cloud.tencent.com/developer/inventory/15332/article/1793670)

手机版“神探玺洛克”请扫码

